Home
Releases
Params
Notes
Code
Download
Home
Releases
Params
Notes
Code
Download
Notes 2024/10/01: fragment ion indexing support in Comet
See notes/2026010_FI for new/updated run stats on updated Comet-FI.
Fragment ion indexing was first introduced by MSFragger in 2017 and this strategy has since been adopted in search tools like MetaMorpheus and Sage. And yes, you are encouraged to go use MSFragger, MetaMorpheus, Sage and all of the other great peptide identification tools out there.
Fragment ion indexing (abbreviated as “FI” or “Comet-FI” going forward) is supported in Comet as of version 2024.02 rev. 0. Given this is the first Comet release with FI functionality, we expect to improve on features, performance, and functionality going forward.
Comet-FI preprint:https://www.biorxiv.org/content/10.1101/2024.10.11.617953v1
Running a Comet-FI search
A Comet-FI search is invoked when the search database is an .idx file. It can be specified in the “database_name” parameter entry in comet.params or via the “-D” command line option. Effectively it’s as simple as specifying “human.fasta.idx” as your search database if the corresponding FASTA file is “human.fasta”.
An .idx file is a plain peptide file containing a list of peptides, their unmodified masses, pointers to proteins that the peptides are present in, and combinatorial bitmasks representing potential variable modification positions. A plain peptide .idx file can be created using the “-i” command line option if you would like to create it directly without it being part of a search. In the first example below, an .idx file will be created for whatever the search database is specified in the comet.params file. In the second example below, the search database “human.fasta” is specified by the command line option “-D” which will override the database specified in comet.params. The new .idx file will be created with the same name as the input FASTA file but with an .idx extension. In the second example below, “human.fasta.idx” would be created.
comet.exe -i
comet.exe -Dhuman.fasta -i
Note that you are also able to simply specify an .idx file as the search database without creating it first. If an .idx database is specified but doesn’t exist, Comet will create the .idx file from the corresponding FASTA file and then proceed with the search.
The examples below all specify the .idx file on the command line using the -D command line option.
comet.exe -Dhuman.fasta.idx somefile.raw
comet.exe -Dhuman.fasta.idx *.mzML
comet.exe -Dhuman.fasta.idx 202410*.mzXML
The commands below would be the equivalent FI search if “database_name = human.fasta.idx” was set in comet.params:
comet.exe somefile.raw
comet.exe *.mzML
comet.exe 202410*.mzXML
Any time the set of variable modifications or the digestion mass range are changed (and I’m sure other parameters I’m forgetting right now), you should re-create the .idx file. If these parameters do not change, you can use the same .idx file. This process of checking if the .idx file needs to be updated will be performed automatically in some future update.
Once a Comet-FI search is invoked, the plain peptide file is parsed, all peptide modification permutations are generated, the bazillion fragment ions calculated and the FI populated. Then the input files are queried against the FI. If multiple input files are searched (aka “comet *.raw”) then the one time cost of generating the FI, which happens once at the beginning of the search, can be avoided for all subsequent files being searched. The process of calculating all fragment ions and populating the index can take a long time and consume lots of memory for search spaces (like large databases, unspecific cleavage rules, and multiple variable modifications).
Current limitations and known issues with Comet-FI:
- MSFragger’s database slicing has not yet been implemented so you must have enough RAM to stored the entire FI in memory. Note that for the real-time search application for intelligent instrument control, database slicing is not feasible.
- Protein n-term and c-term variable modifications are not supported in this initial FI release. This functionality is expected to be added soon. This means that variable modifications are limited to residues and peptide termini.
- Only variable_mod01 through variable_mod05 are supported with FI. This is a limit imposed to restrict the FI to a reasonable size.
- For each variable_modXX, a maximum of 5 modified residues will be considered in a peptide. This might further be limited by the total allowed number of modified residues in a peptide controlled by the max_variable_mods_in_peptide parameter.
- Comet’s internal decoy search via the decoy_search parameter is not supported. For FDR analysis, you should supply Comet a FASTA file containing target and decoy entries.
- It’s simply not in your best interest to try running this with many variable modifications combined with a no-enzyme constraint and a large database; you’re better served using another search engine instead. Any search requiring over 100 GB of RAM should not be attempted.
Fragment ion index specific search parameters
- fragindex_min_fragmentmass
- fragindex_max_fragmentmass
- fragindex_min_ions_report
- fragindex_min_ions_score
- fragindex_num_spectrumpeaks
- fragindex_skipreadprecursors
Memory use and performance
There are many factors that go into how much memory will be consumed including:
- database size
- peptide length range
- enzyme constraints including missed cleavages and semi or nonspecific cleavages
- number of variable modifications considered
- mass range of the fragment ions used in the index
One can easily generate over a billion fragment ions in a standard human, target + decoy, tryptic analysis by adding a few variable modifications. And representing a billion fragment ions in a fragment index in memory will require many GBs of RAM. You might get away with some smaller searches on a 16 GB or 32 GB machine. Many searches can be done with 64 GB RAM. And if you’re a power user who wants to analyze MHC peptides requiring non-specific enzyme constraint searches, don’t attempt to run such a search with many modifications as it simply won’t work.
The following searches were run using 8-cores of an AMD Epyc 7443P processor running on Ubuntu linux version 22.04. The query file is a two hour Orbitrap Lumos run with MS/MS spectra acquired in high res (approximately 92,000 MS/MS spectra). Up to two of each specified variable modifications are allowed in a peptide, peptide length 7 to 50, digest mass range 600.0 to 5000.0, tryptic digest with 2 allowed missed cleavages, +/- 20 ppm precursor tolerance considering up to two isotope offsets. The .idx files were created before the search so the reported search times and .idx creation times are both noted.
- Yeast forward + reverse (12,488 sequence entries), variable mods 16M and 80STY uses 5.6 GB of RAM and completes in 0:30. The plain peptide .idx file creation step took 0:03 to complete.
- Human forward + reverse (193,864 sequence entries), variable mods 16M uses 5.2 GB of RAM and completes in 0:31. During the search, populating the fragment ion index took 0:11 and the search took 0:20. The .idx creation step took 0:22 to generate.
- Human forward + reverse, tryptic, variable mods 16M and 80STY uses 11.3 GB of RAM and completes in 1:08. Corresponding standard Comet search took 4:10.
- Human forward + reverse, tryptic, variable mods 16M, 80STY, 229nK, 42K, and 0.98c (allow 1 of each modification except for the TMT label where up to 2 mods are allowed; 2 total modifications allowed on a peptide) uses 74 GB of RAM and completes in 7:34. The vast majority of that time is in generation the fragment ion index as the actual search took 0:29. The .idx creation step took 0:38 to generate and the corresponding standard Comet search took 28:07.
- Human forward + reverse sequences, semi-tryptic enzyme constraint, 16M and 80ST variable mods uses 103 GB of RAM and completes in 11:06. Note that every additional file searched (aka comet *.mzML) would add an extra ~50 seconds per file to complete so there’s benefits to giving Comet-FI multiple files to search in order to amortize the cost of the fragment ion index creation across all query files. The .idx creation step took 7:14 to generate and consumed 52 GB of RAM. The corresponding standard Comet search took 40:46.
- Human forward + reverse sequences, no enzyme constraint, no variable mods, peptide length range 7 to 15 uses 45 GB of RAM and completes in 4:45. Most of that time is for the fragment ion index creation as the actual search took just 0:36. The corresponding standard Comet search took 15:48. It’s worth noting that the .idx creation step consumed a whopping 105 GB RAM and took 11:42.
- Same human forward + reverse sequences, no enzyme constraint, peptide length range 7 to 15 but adding in 16M variable mod uses 49 GB of RAM and completes in 5:30. The corresponding standard Comet search took 20:05. The .idx creation step consumed 106 GB RAM and took 12:40.
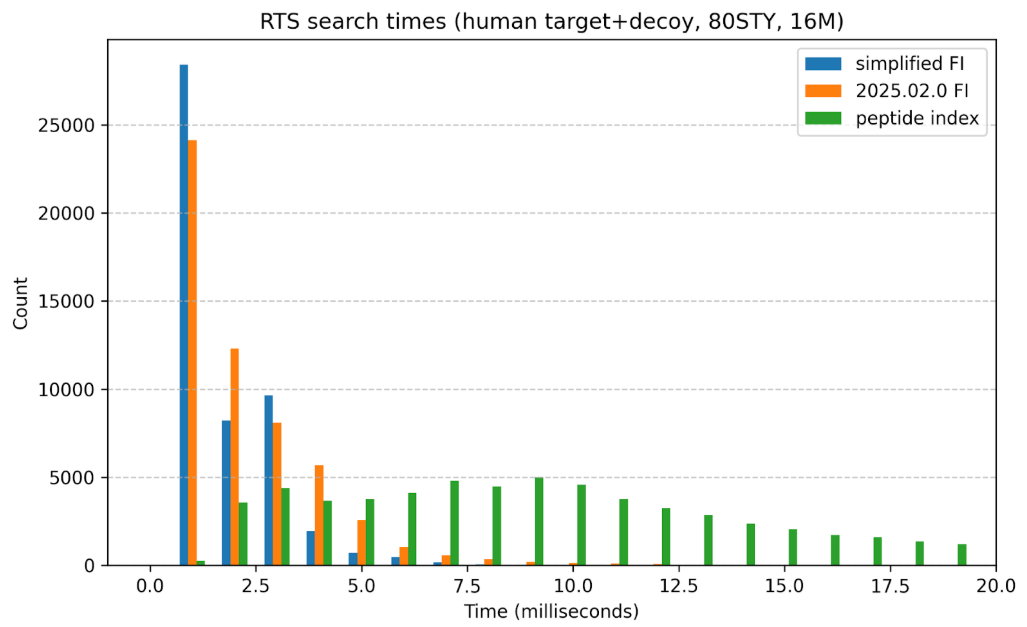
2025/11/12 simplied fragment ion index
With release v2025.03.0,the fragment ion index was simplified, removing thread and precursor mass range dimensions. This greatly simplified the index but didn’t significantly affect the performance (search times).
Although I see search times vary, based on different compilations of the code and when I run the timings, this plot shows one example comparing the fragment ion index search performance for release 2025.03.0 vs. 2025.02.0 vs. the old peptide indexing.